Non, BD continuera à utiliser des numéros de catalogue en plus des numéros d’articles commerciaux internationaux GS1 (GTIN) pour l’identification des dispositifs.
true
Distributeurs
BD fabrique et vend une vaste gamme de produits et services pour les marchés des soins de santé, du grand public, des sciences de la vie et de l’industrie. La plupart de nos produits sont vendus par l’intermédiaire de distributeurs et de grossistes.

- Liste des distributeurs
- Identifiant unique du dispositif (UDI)
Conformité à la réglementation sur l’UDI
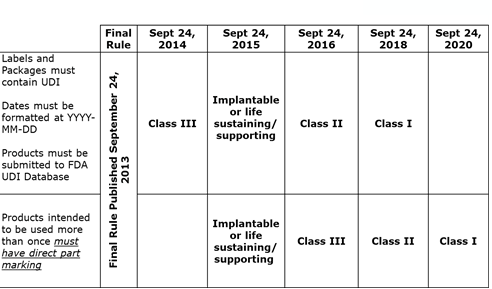
Le 24 septembre 2013, la Food and Drug Administration (FDA) des États-Unis a publié une règle finale exigeant que la plupart des dispositifs médicaux distribués aux États-Unis portent un identifiant unique de dispositif (UDI). Le système UDI facilite l’identification, la traçabilité et le suivi des dispositifs médicaux tout au long de leur distribution et de leur utilisation.
Le système UDI se compose de trois segments fondamentaux. Le premier est l’UDI, un numéro unique attribué à la version ou au modèle d’un dispositif requis sur l’étiquetage d’emballage des produits finis. L’UDI doit inclure un identificateur de dispositif (DI) et peut contenir un identificateur de production (PI). Les UDI apparaissent sur les étiquettes au format texte brut et dans un format lisible par la technologie de capture automatique des données d’identification (AIDC) (p. ex. un code-barres). Cet identifiant comprend également des informations spécifiques à la production, comme le numéro de lot du produit, la date de péremption et la date de fabrication à laquelle ces informations apparaissent sur l’étiquette. En outre, la date doit être au format « AAAA-MM-JJ » sur tout texte lisible par l’homme (qui n’inclut pas le texte du code à barres).
Le deuxième composant est le marquage direct des pièces, qui est un marquage UDI permanent sur le dispositif. Si un dispositif doit être utilisé plusieurs fois et être retraité avant chaque utilisation, il doit également être directement marqué d’un UDI, permettant une identification précise même lorsque le dispositif n’est plus accompagné de son étiquette ou de son emballage.
Le troisième composant est la base de données mondiale d’identification unique des dispositifs (GUDID), qui est une base de données consultable publiquement administrée par la FDA et qui sert de catalogue de référence pour chaque dispositif avec un identifiant.
Chronologie de la conformité UDI
La FDA a défini une approche progressive de la conformité en fonction de la classification des risques du dispositif (reportez-vous au tableau chronologique de l’UDI de la FDA).
BD exige que tous ses fournisseurs mettent en œuvre et respectent le calendrier de l’UDI de la FDA, en répondant rapidement aux questions et en communiquant régulièrement sur les efforts en matière d’UDI.
Visite Identifiant de dispositif unique (UDI) ou AccessGUDID sur le site Web de la FDA, ou communiquez avec votre représentant BD pour plus d’informations sur le système UDI.
CALENDRIER UDI DE LA FDA
×
![udi-timeline.png]()
En 2013, la Food and Drug Administration (FDA) des États-Unis a publié une règle finalela mise en place d’un système unique d’identification des dispositifs conçu pour identifier correctement les dispositifs tout au long de la distribution et de l’utilisation. La règle finale exige que les étiqueteurs du dispositif incluent un identifiant de dispositif unique (UDI) sur les étiquettes et les emballages du dispositif, sauf si la règle prévoit une exception ou une alternative. Chaque UDI doit être fourni dans une version en texte brut et sous un format utilisant une technologie d’identification et de saisie des données automatique (AIDC). L’UDI devra également être directement marqué sur un dispositif destiné à plusieurs usages et à être retraité avant chaque utilisation. Les dates figurant sur les étiquettes et les emballages des dispositifs doivent être présentées dans un format standard conforme aux normes et aux pratiques internationales.
Un UDI est un code numérique ou alphanumérique unique qui se compose des deux parties suivantes :
1. Un identifiant de dispositif (DI), partie fixe obligatoire d’un UDI identifiant l’étiqueteur et la version ou le modèle spécifique d’un dispositif
2. Un identificateur de production (PI), partie conditionnelle et variable d’un UDI qui identifie un ou plusieurs des détails suivants lorsqu’ils sont inclus sur l’étiquette d’un dispositif :
- Le numéro du lot auquel le dispositif appartient
- Le numéro de série d’un dispositif spécifique
- La date de péremption d’un dispositif spécifique
- La date à laquelle un dispositif spécifique a été fabriqué
- Code d’identification distinct exigé par le §1271.290(c) pour une cellule humaine, un tissu ou un produit à base de cellules et tissus (HCT/P) réglementé comme un dispositif1
Cette règle finale réduira considérablement les obstacles existants à l’identification adéquate des dispositifs médicaux utilisés aux États-Unis. En permettant d’identifier rapidement et définitivement un dispositif et les caractéristiques clés qui affectent sa sécurité et son efficacité d’utilisation, la règle permettra de réduire les erreurs médicales qui résultent de la mauvaise identification d’un dispositif ou de la confusion quant à son utilisation appropriée. Le système d’identification mis en place dans le cadre de cette règle permettra d’améliorer la précision de la déclaration des événements indésirables en facilitant l’identification du dispositif avant l’envoi d’un rapport. Elle permettra à la FDA, aux prestataires de soins de santé et au secteur d’extraire plus rapidement des informations utiles à partir des rapports d’événements indésirables, d’identifier le dispositif en question et, par conséquent, de mieux comprendre les problèmes sous-jacents et de prendre des mesures correctives appropriées, mieux focalisées. La règle exige également que les dates indiquées sur les étiquettes des dispositifs médicaux soient conformes à un format standard, afin de s’assurer que ces dates sont sans ambiguïté et clairement comprises par les utilisateurs du dispositif.
En 2013, la Food and Drug Administration (FDA) des États-Unis a publié un règle finale établissant un système unique d’identification des dispositifs, conçu pour identifier correctement les dispositifs tout au long de leur distribution et de leur utilisation. La règle finale exige que les étiqueteurs du dispositif incluent un identifiant de dispositif unique (UDI) sur les étiquettes et les emballages du dispositif, sauf si la règle prévoit une exception ou une alternative. Chaque UDI doit être fourni dans une version en texte brut et sous un format utilisant une technologie d’identification et de saisie des données automatique (AIDC). L’UDI devra également être directement marqué sur un dispositif destiné à plusieurs usages et à être retraité avant chaque utilisation.
Oui. Les processus de commande de BD continueront d’utiliser les références catalogue BD actuelles; le processus de commande restera inchangé si vous préférez commander par référence catalogue BD.
Si vous souhaitez utiliser le DI (GTIN) pour vos processus de commande par échange de données informatisées (EDI), contactez votre représentant BD.
Oui. En plus d’utiliser le numéro de catalogue BD, BD a commencé à améliorer ses factures ou ses bordereaux d’expédition pour inclure à la fois les renseignements DI et PI.
Communiquez avec votre représentant BD pour obtenir des renseignements spécifiques ou consultez le site AccessGUDID.
BD fabrique toutes les classes de dispositifs médicaux. Nous fabriquons également des produits exemptés de la réglementation sur l’UDI de la FDA, tels que les produits pharmaceutiques et les produits destinés à la recherche uniquement (RUO).
Oui. BD a déjà commencé le processus de renseignement de la base de données gUDID (Global Unique Device Identification Database) de la FDA, un référentiel d’informations sur les produits. Les produits BD de classe III sont déjà dans la base GUDID. D’autres produits seront ajoutés à la base GUDID sur ou avant le calendrier UDI de la FDA.
D’une manière générale, vous constaterez que les nouvelles étiquettes BD sont améliorées avec des données supplémentaires et continueront à fonctionner dans vos processus de scan. Vous serez informé(e) de tout changement de DI ou de PI (et de numéro de catalogue et de code-barres associés) suite à la mise en œuvre de l’UDI de BD.
true